10.4 Full taxonomy annotation
In microbial data analysis, the data are often collapsed into different levels of Phylum, Order, Class, Family, Genus and Species for downstream analysis. However, due to the limitation of database annotations, some taxa may have lower-level annotations with the same name but upper-level annotations with different names. This situation will cause the EMP_collapse function to merge different bacteria in genus-level data collapse incorrectly. As follows:
| Family | Genus | Species |
|---|---|---|
| Lachnospiraceae | Ruminococcus | unclassified |
| Ruminococcaceae | Ruminococcus | unclassified |
| Oscillospiraceae | Ruminococcus | unclassified |
Therefore, EasyMultiProfiler provides two microbial annotation methods: complete-level annotation and single-level annotation. Users can choose the appropriate method according to their needs. 1. Full-level annotation is more rigorous in annotation and is suitable for analyses that do not involve specific bacterial names (such as alpha diversity, and beta diversity analysis). In this case, using the complete level annotation is recommended. If a drawing involving specific bacterial names is involved, it is recommended to use ggsave to save the graph first, and then manually adjust the labels and annotations to improve the readability of the graph. 2. Single-level annotation can meet the needs in most cases and will not affect the visualization effect.
🏷️Example1:
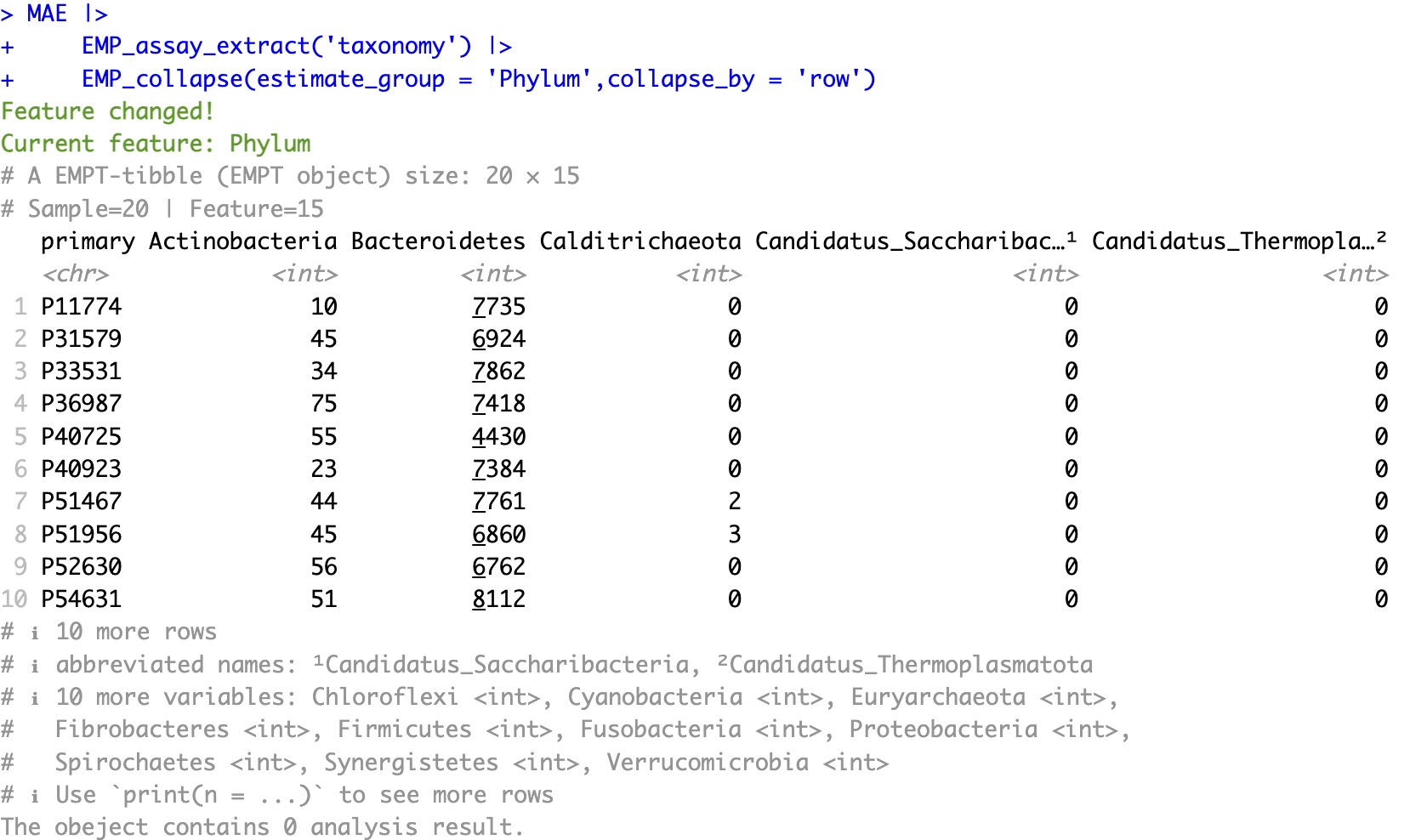
The original feature is a single-level annotation:
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_collapse(estimate_group = 'Phylum',collapse_by = 'row')
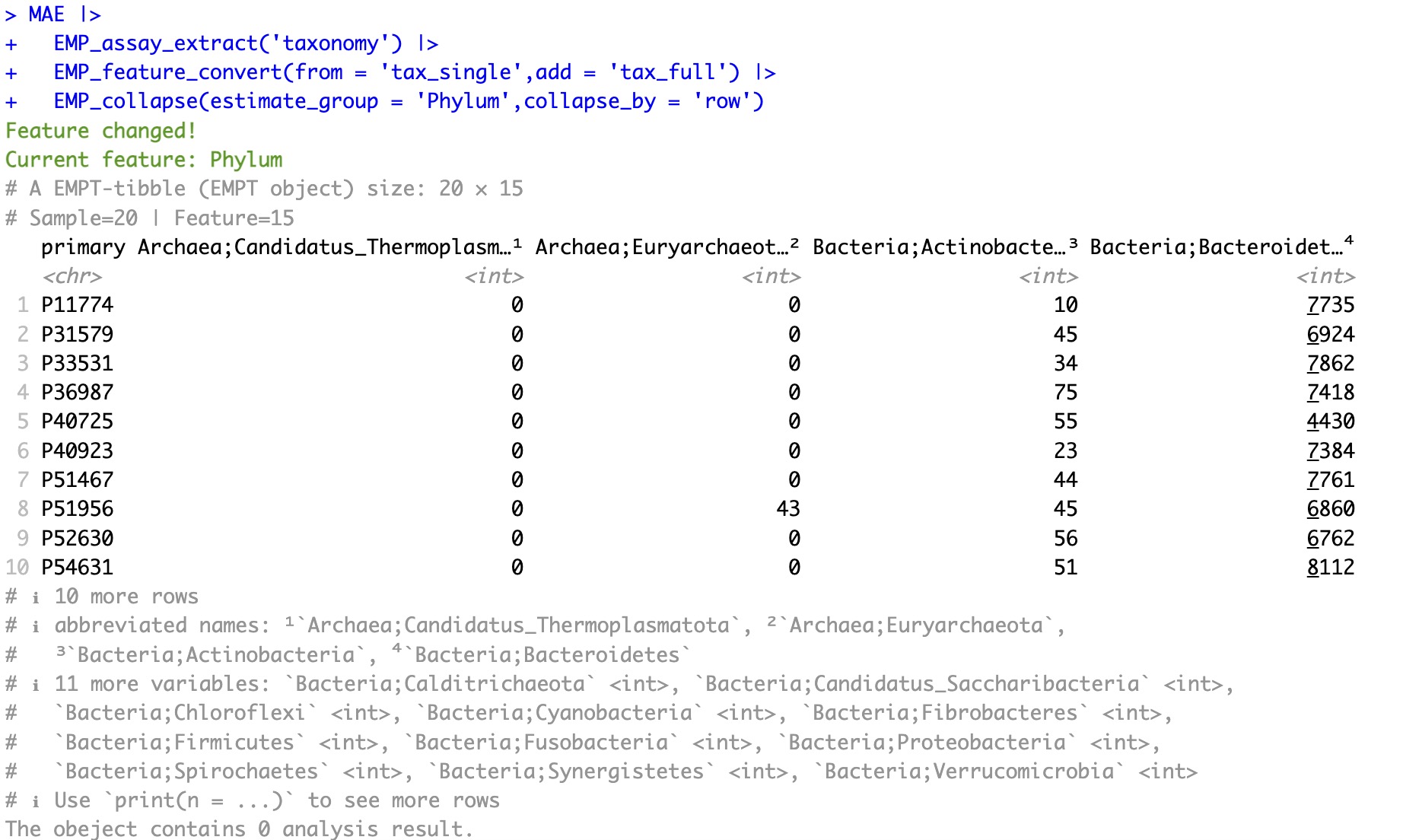
Convert to complete-level annotation:
This function can only work before collpsing microbial data based on taxonomy profile.
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_feature_convert(from = 'tax_single',add = 'tax_full') |>
EMP_collapse(estimate_group = 'Phylum',collapse_by = 'row')
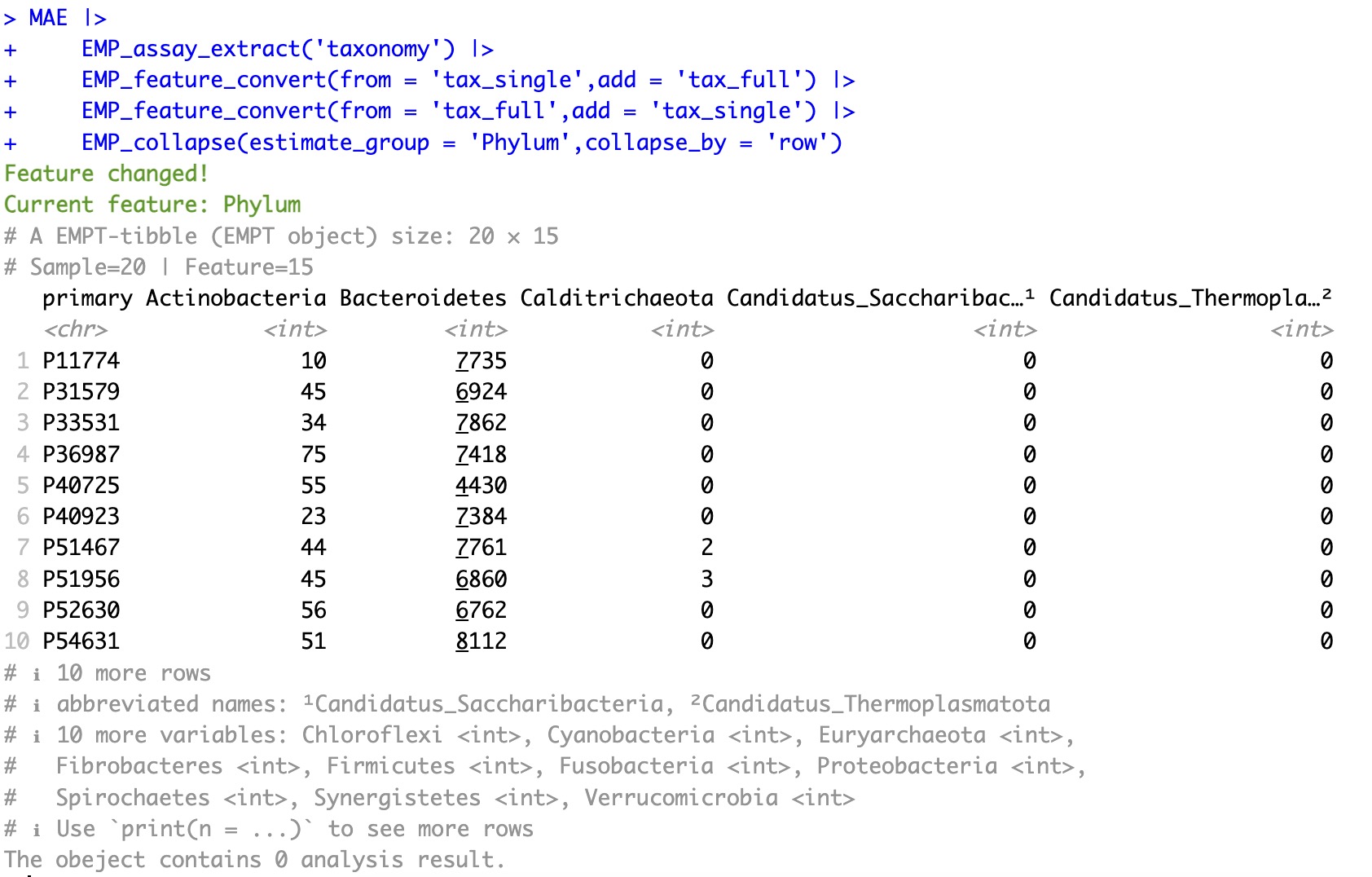
Covert complete-level annotation back to single-level annotation:
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_feature_convert(from = 'tax_single',add = 'tax_full') |>
EMP_feature_convert(from = 'tax_full',add = 'tax_single') |>
EMP_collapse(estimate_group = 'Phylum',collapse_by = 'row')
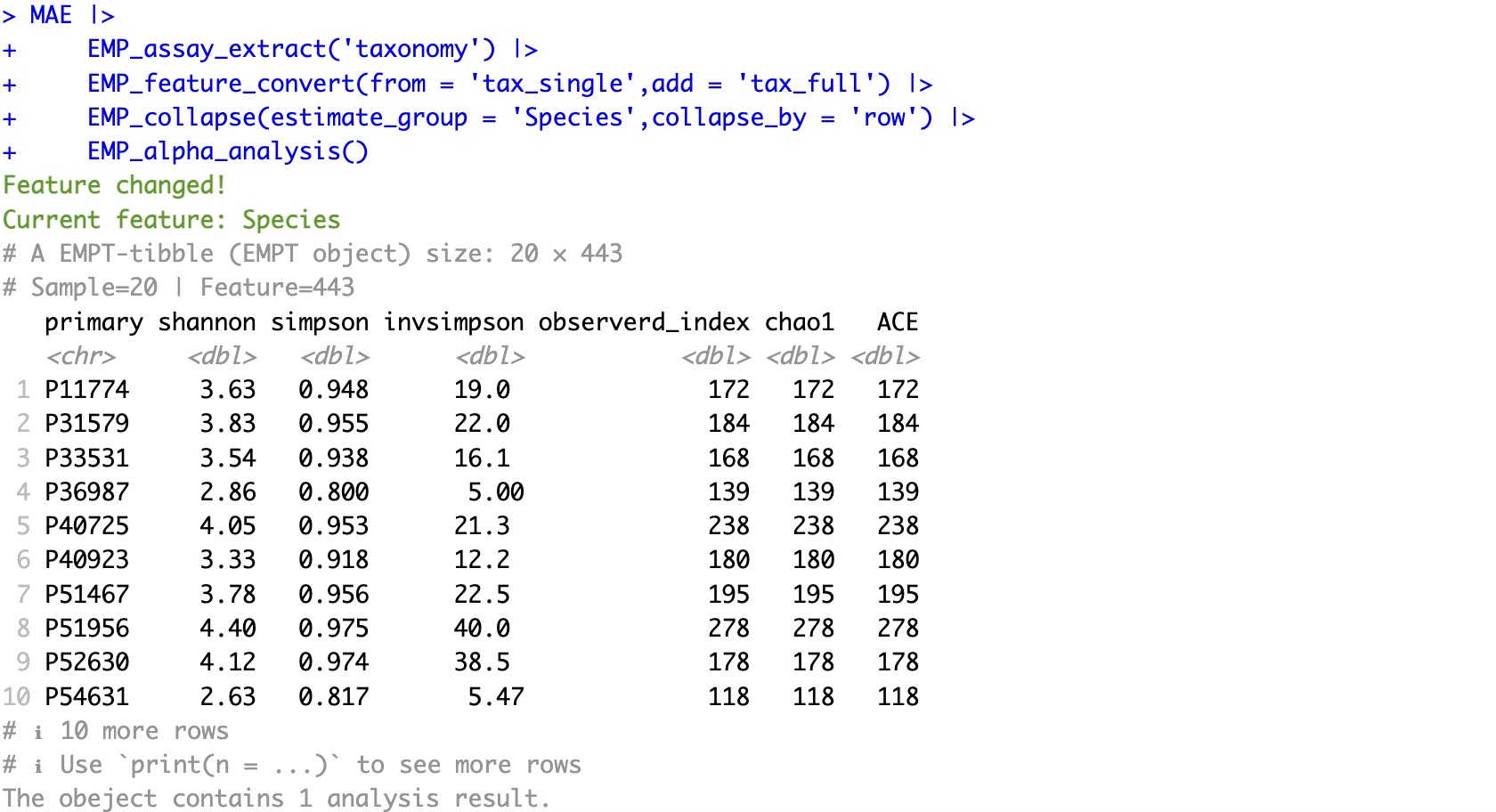
🏷️Example2: Diversity analysis after complete annotation
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_feature_convert(from = 'tax_single',add = 'tax_full') |>
EMP_collapse(estimate_group = 'Species',collapse_by = 'row') |>
EMP_alpha_analysis()