10.6 Microbiome Single-Omics Quick-Start Example
This section demonstrates a complete and commonly used analytical workflow for microbiome single-omics data using example datasets.
Example data download: Github link
10.6.1 Importing Microbiome Data
For single-omic data where no relationship table is involved, the sampleID in the abundance matrix must match those in the Sample phenotypic data.
library(EasyMultiProfiler)
meta_data <- read.table('coldata.txt',header = T,row.names = 1)
data <- read.table('tax.txt',header = T,sep = '\t')
MAE <- EMP_easy_import(data = data,coldata = meta_data,type = 'tax')
10.6.2 Exploring Microbiome Data
View Current Microbiome Assay
MAE |>
EMP_assay_extract() # View expression matrix
MAE |>
EMP_coldata_extract() # View phenotype data
MAE |>
EMP_rowdata_extract() # View taxonomic annotations
View Species-Level Data
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Species')
View Class-Level Data
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Class')
View Phylum-Level Data
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Phylum') |>
EMP_structure_plot(top_num = 10)
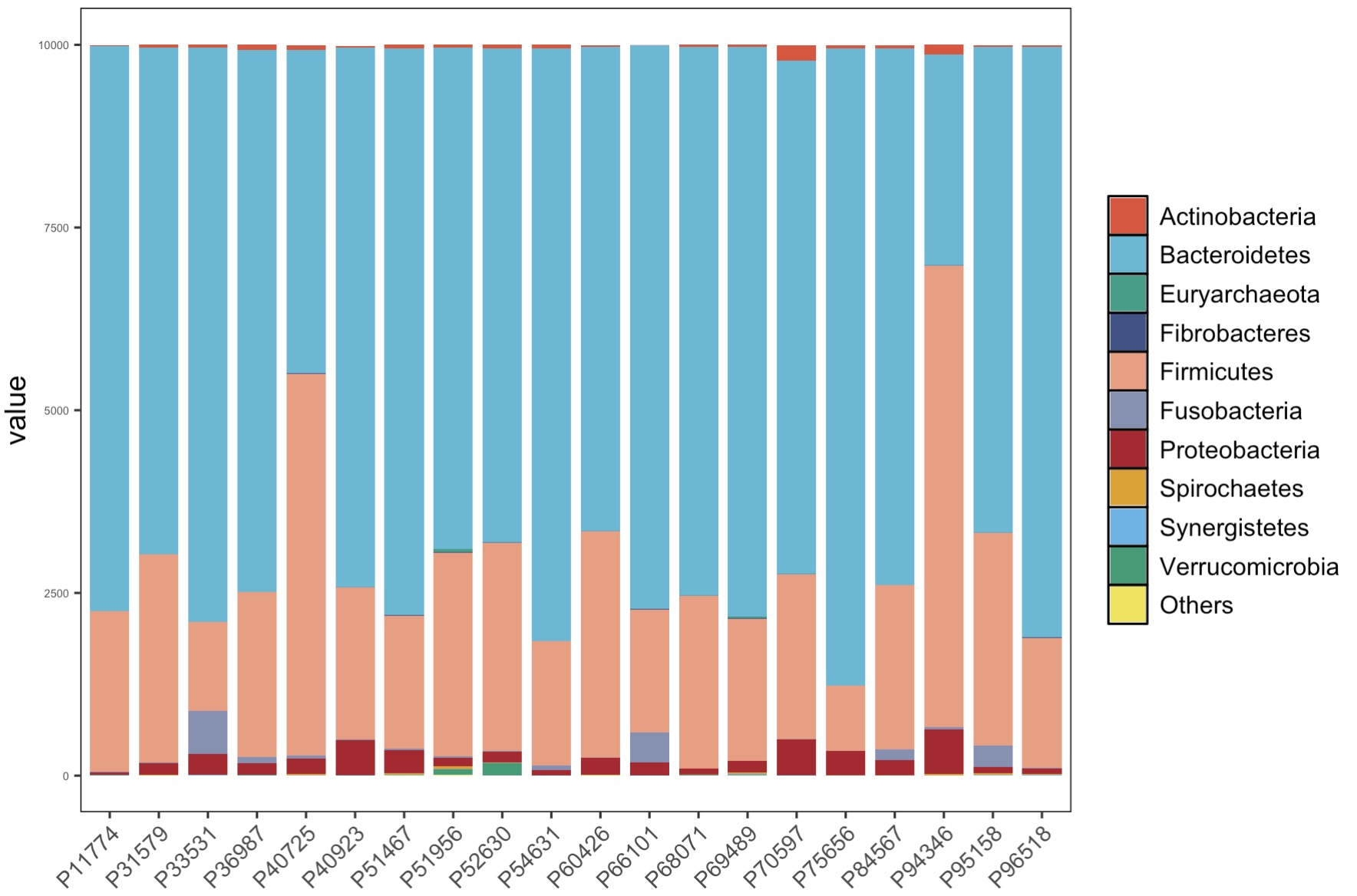
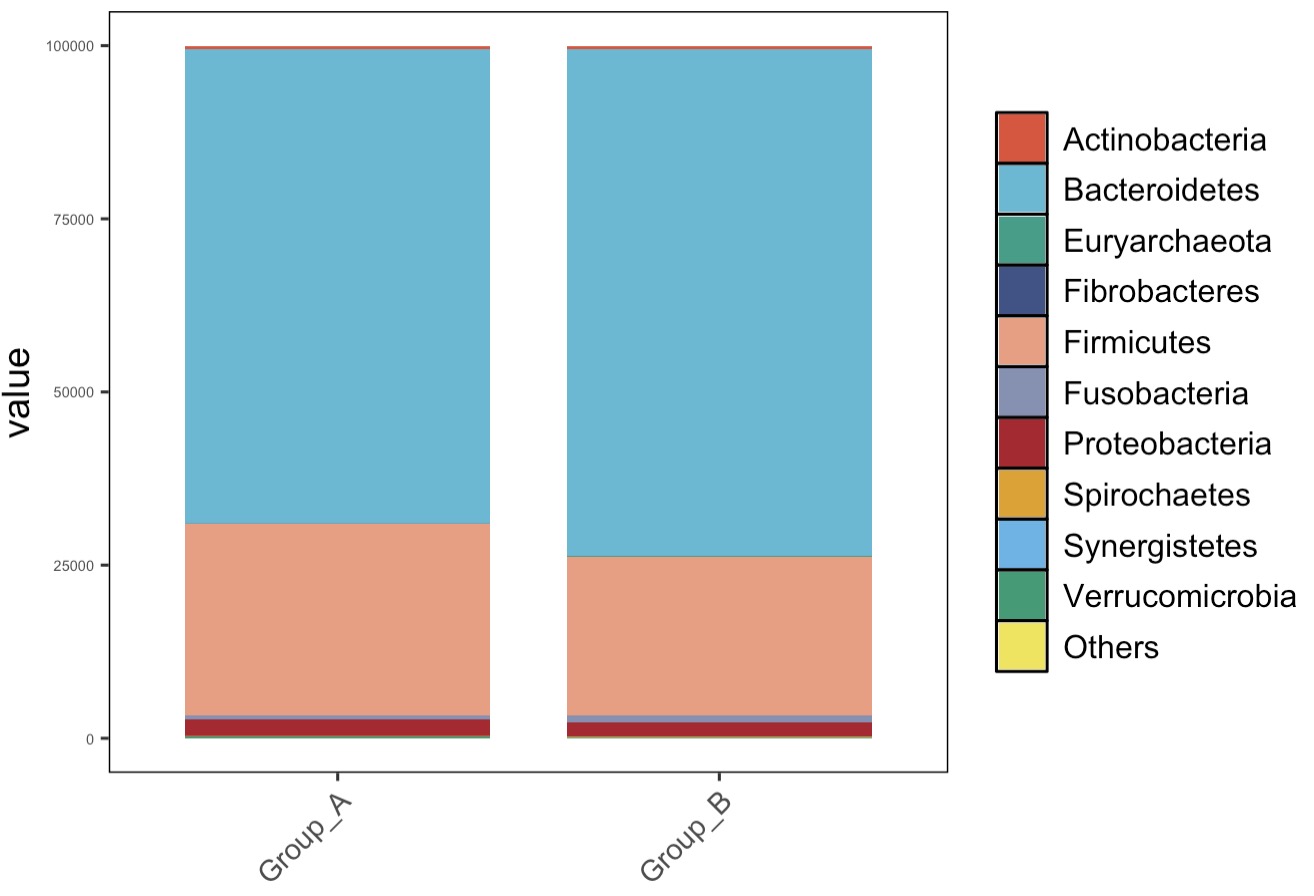
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Phylum') |>
EMP_collapse(collapse_by = 'col',estimate_group = 'Group') |>
EMP_structure_plot(top_num = 10)
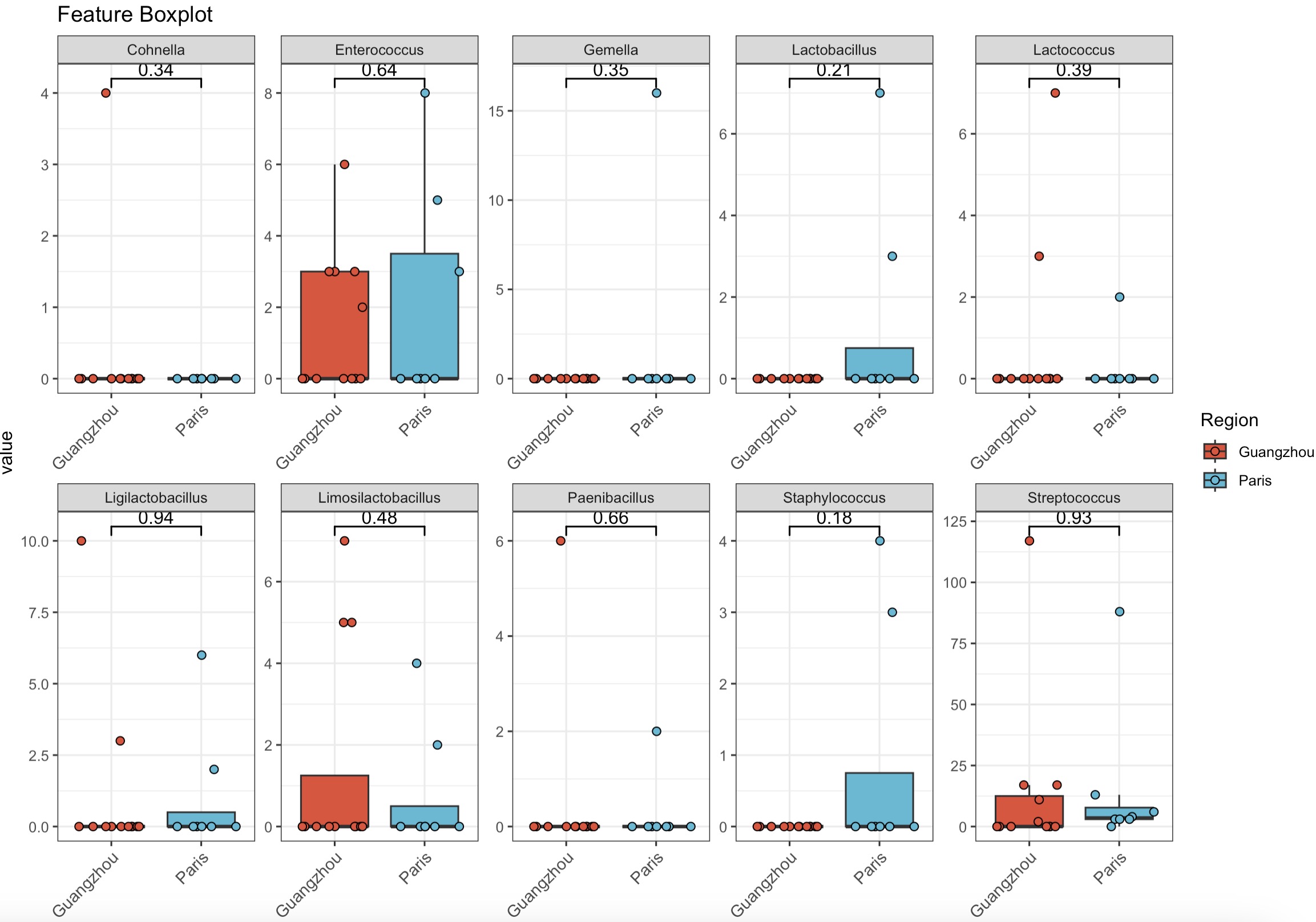
Examine the bacterial genera within the class Bacilli and perform statistical tests with the data grouped by region
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_filter(feature_condition = Class %in% 'Bacilli') |>
EMP_boxplot(estimate_group='Region',
method='t.test',
ncol=5)
10.6.3 Rarefaction (Optional)
Rarefy Using the Smallest Read Count Among Samples
MAE |>
EMP_assay_extract() |>
EMP_rrarefy()
Rarefy with a Custom Minimum Read Count
MAE |>
EMP_assay_extract() |>
EMP_rrarefy(raresize = 5000)
10.6.4 Data Normalization
Convert to Relative Abundance at Genus Level
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_decostand(method = 'relative')
Apply CLR Transformation at Genus Level
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_decostand(method = 'clr')
Apply Log2 Transformation at Genus Level
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_decostand(method = 'log2+1')
10.6.5 Batch Effect Correction (Optional)
Correct for Batch Effects by Region at Genus Level
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_adjust_abundance(.factor_unwanted = 'Region',
.factor_of_interest = 'Group',
method = 'combat_seq')
10.6.6 Core Microbiome Identification
Identify Core Genera with Minimum Abundance 0.001 and Prevalence >70% in at Least One Group
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7)
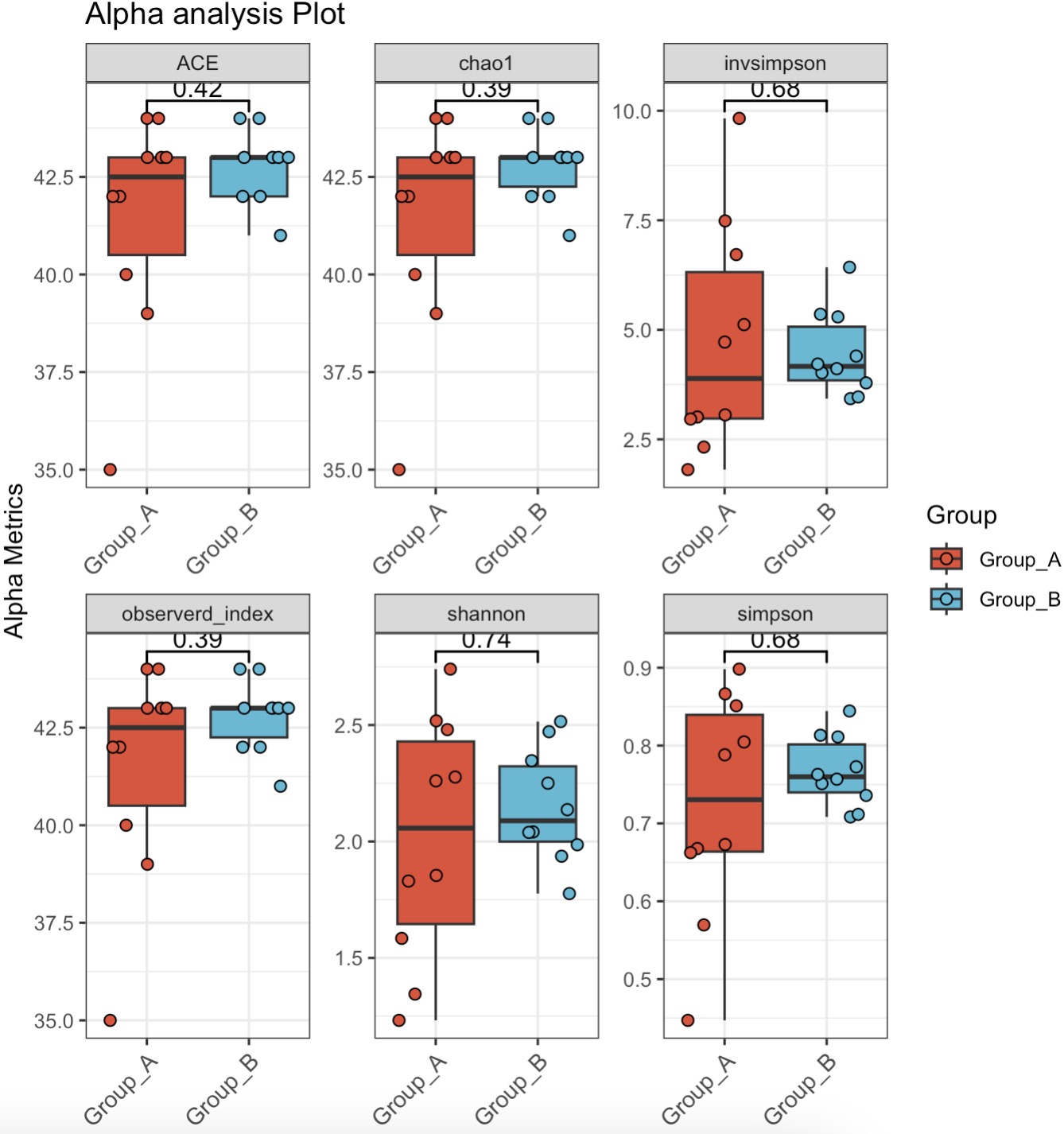
10.6.7 Alpha Diversity Analysis
Calculate Alpha Diversity for Core Genera and Visualize with Boxplot
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7) |>
EMP_alpha_analysis() |>
EMP_boxplot(estimate_group = 'Group')
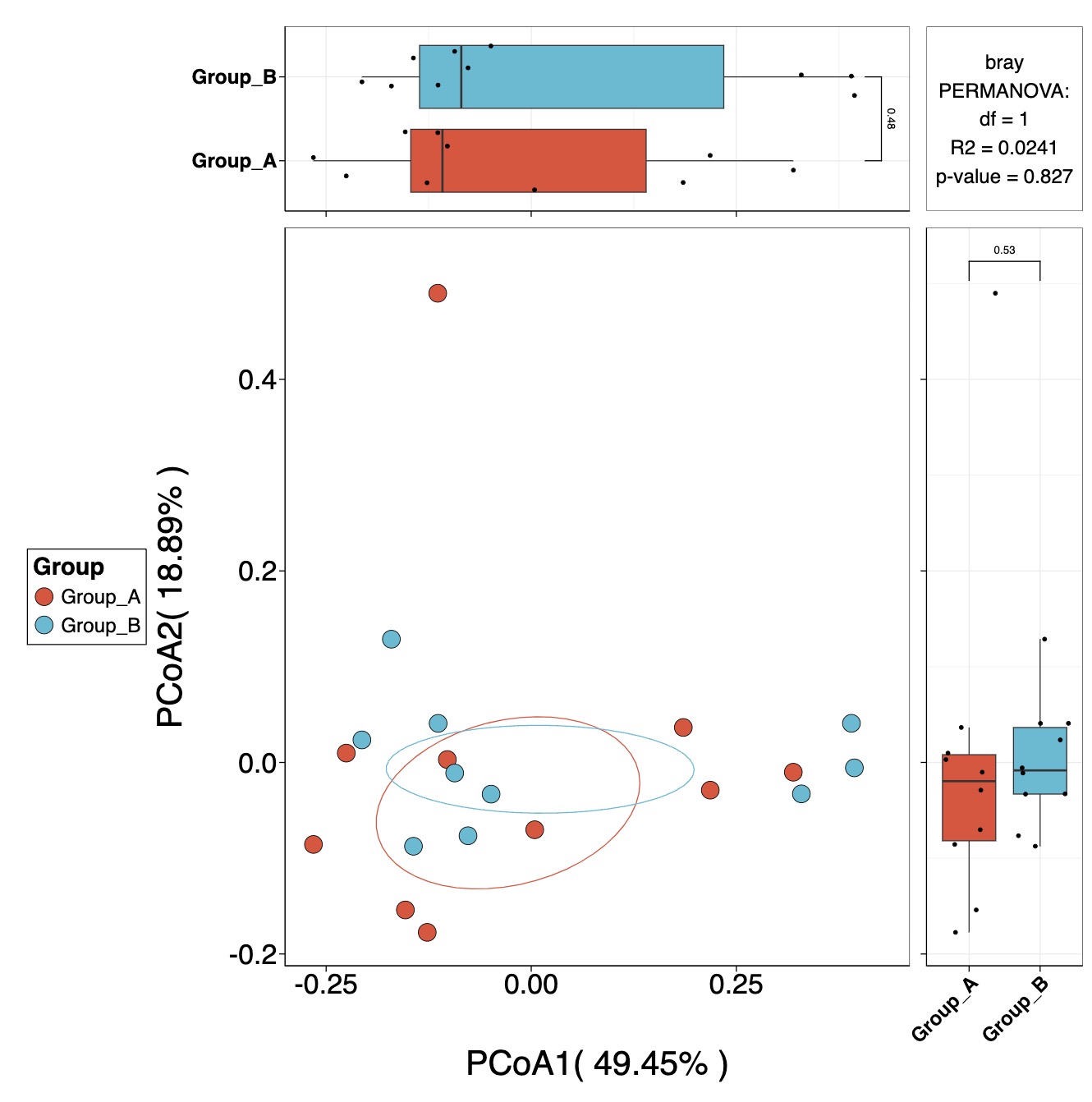
10.6.8 Beta Diversity Analysis
Calculate Beta Diversity for Core Genera and Generate Ordination Plot
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7) |>
EMP_dimension_analysis(method = 'pcoa',distance = 'bray') |>
EMP_scatterplot(estimate_group = 'Group',show='p12html')
10.6.9 Differential Abundance Analysis
Perform Wilcoxon Test and Filter Significant Genera
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group') |>
EMP_filter(feature_condition = pvalue < 0.05)
Perform DESeq2 Analysis and Filter Significant Genera
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_filter(feature_condition = pvalue < 0.05,keep_result = TRUE)
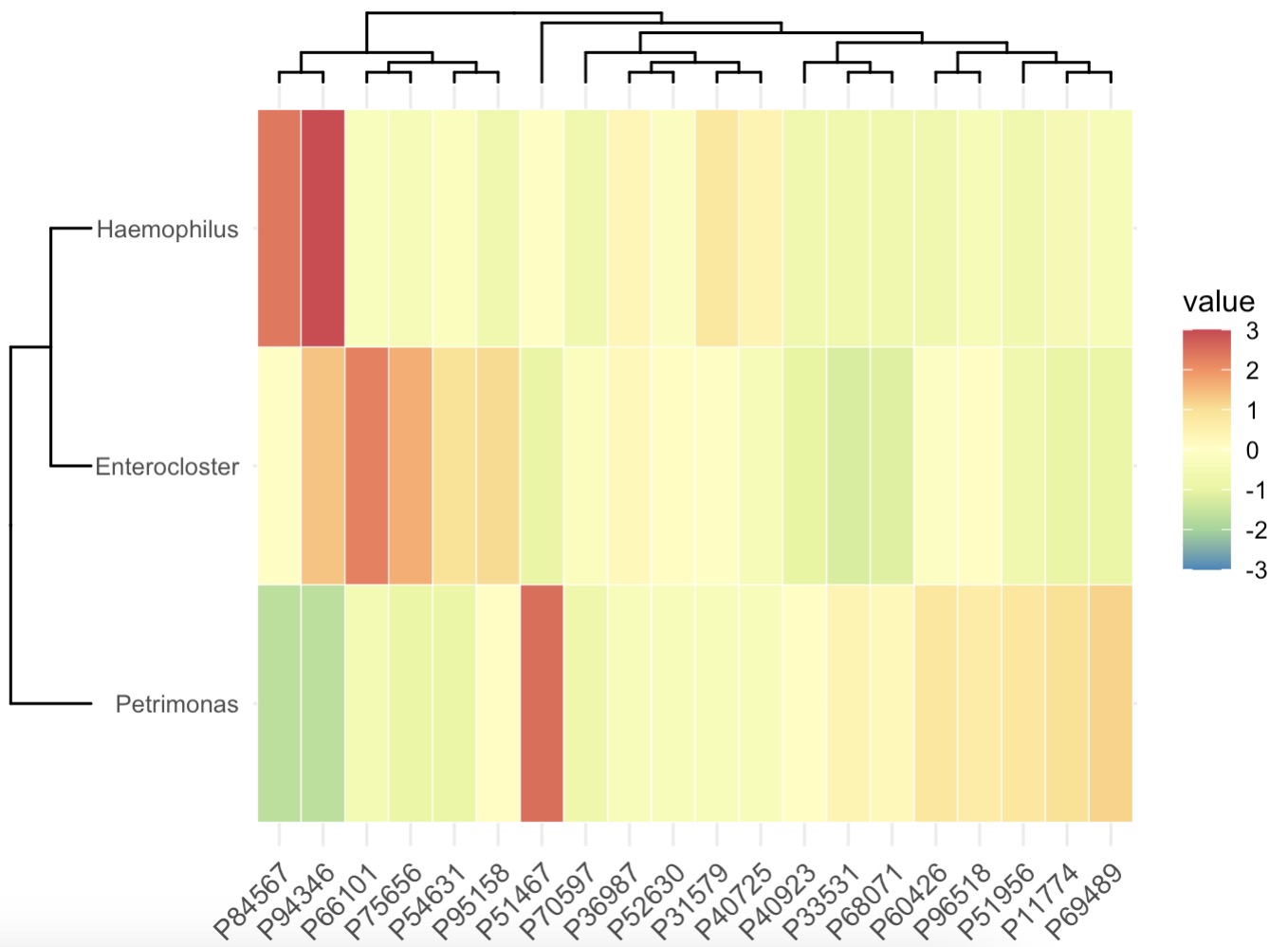
10.6.10 Machine Learning for Key Taxa
The EMP package includes built-in methods for feature selection: Boruta, Random Forest, XGBoost, and Lasso. For detailed usage, run help(EMP_marker_analysis).
Identify Important Genera Using Boruta and Visualize with Heatmap
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7) |>
EMP_marker_analysis(method = 'boruta',estimate_group = 'Group') |>
EMP_filter(feature_condition = Boruta_decision!= 'Rejected') |>
EMP_heatmap_plot(palette='Spectral',legend_bar='auto',
clust_row=TRUE,clust_col=TRUE)
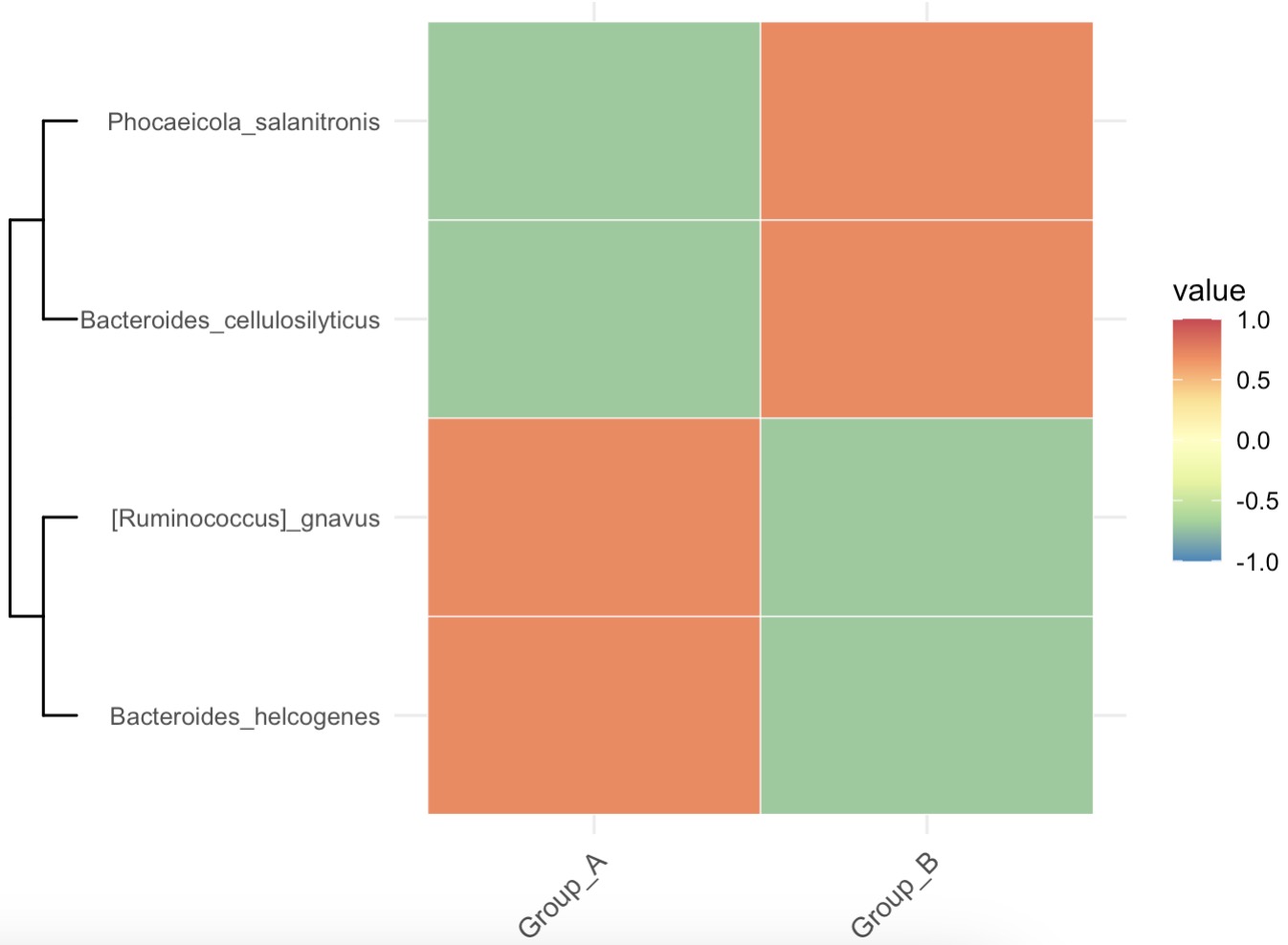
Select Height-Associated Genera Using Lasso and Plot Group-Level Heatmap
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Species') |>
EMP_identify_assay(estimate_group = 'Group',method = 'default',
min=0.001,min_ratio = 0.7) |>
EMP_marker_analysis(method = 'lasso',estimate_group = 'Height') |>
EMP_filter(feature_condition = lasso_coe > 0) |>
EMP_collapse(method = 'mean',estimate_group = 'Group',
collapse_by = 'col') |>
EMP_heatmap_plot(palette='Spectral',legend_bar='auto')
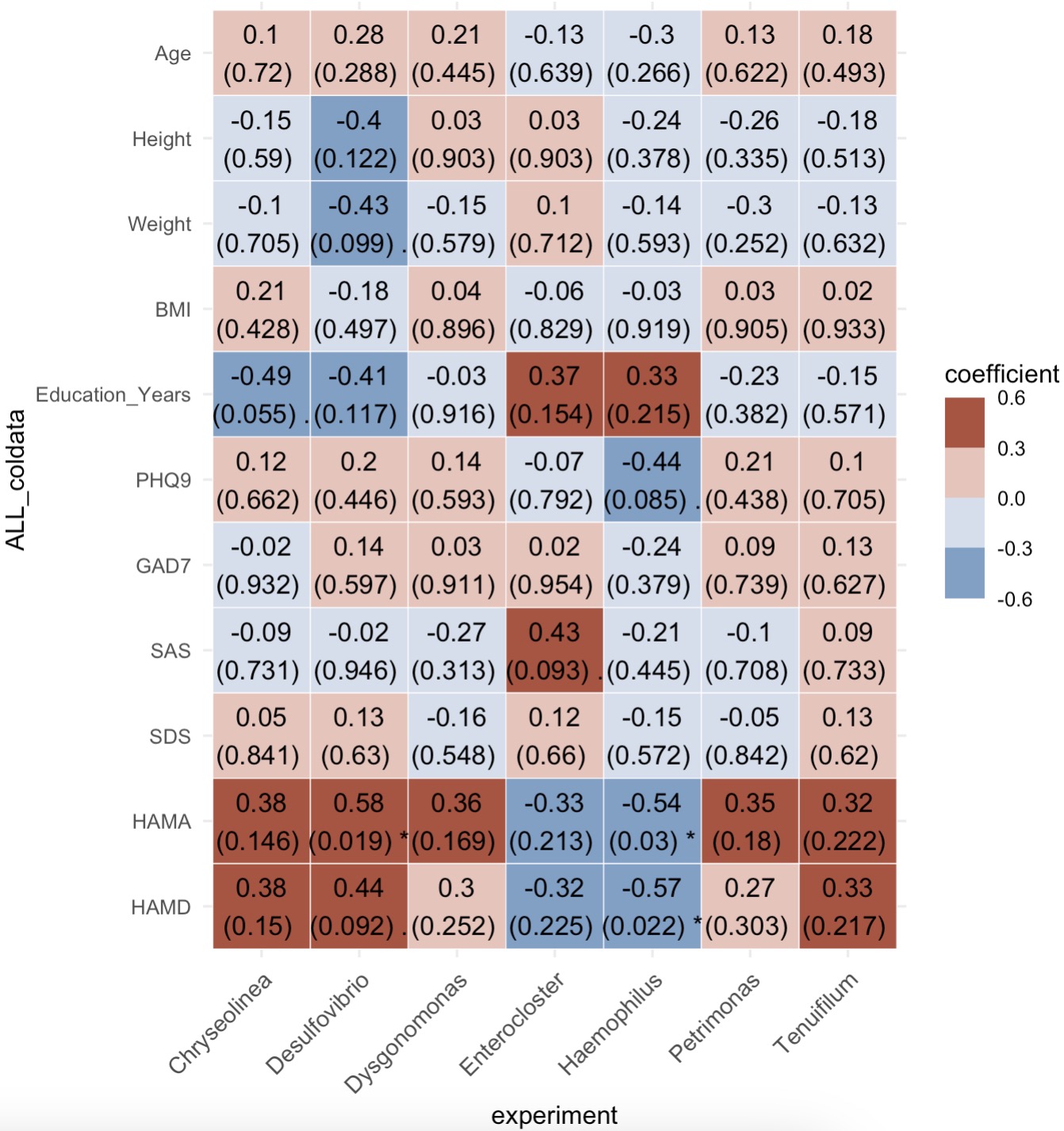
10.6.11 Correlation with Phenotypes
Correlation Heatmap Between diff genus and Phenotypic Variables
diff_genus <- MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group') |>
EMP_filter(feature_condition = pvalue < 0.05)
meta_data <- MAE |>
EMP_coldata_extract(action = 'add')
(phylum_data + meta_data) |>
EMP_cor_analysis() |>
EMP_heatmap_plot()
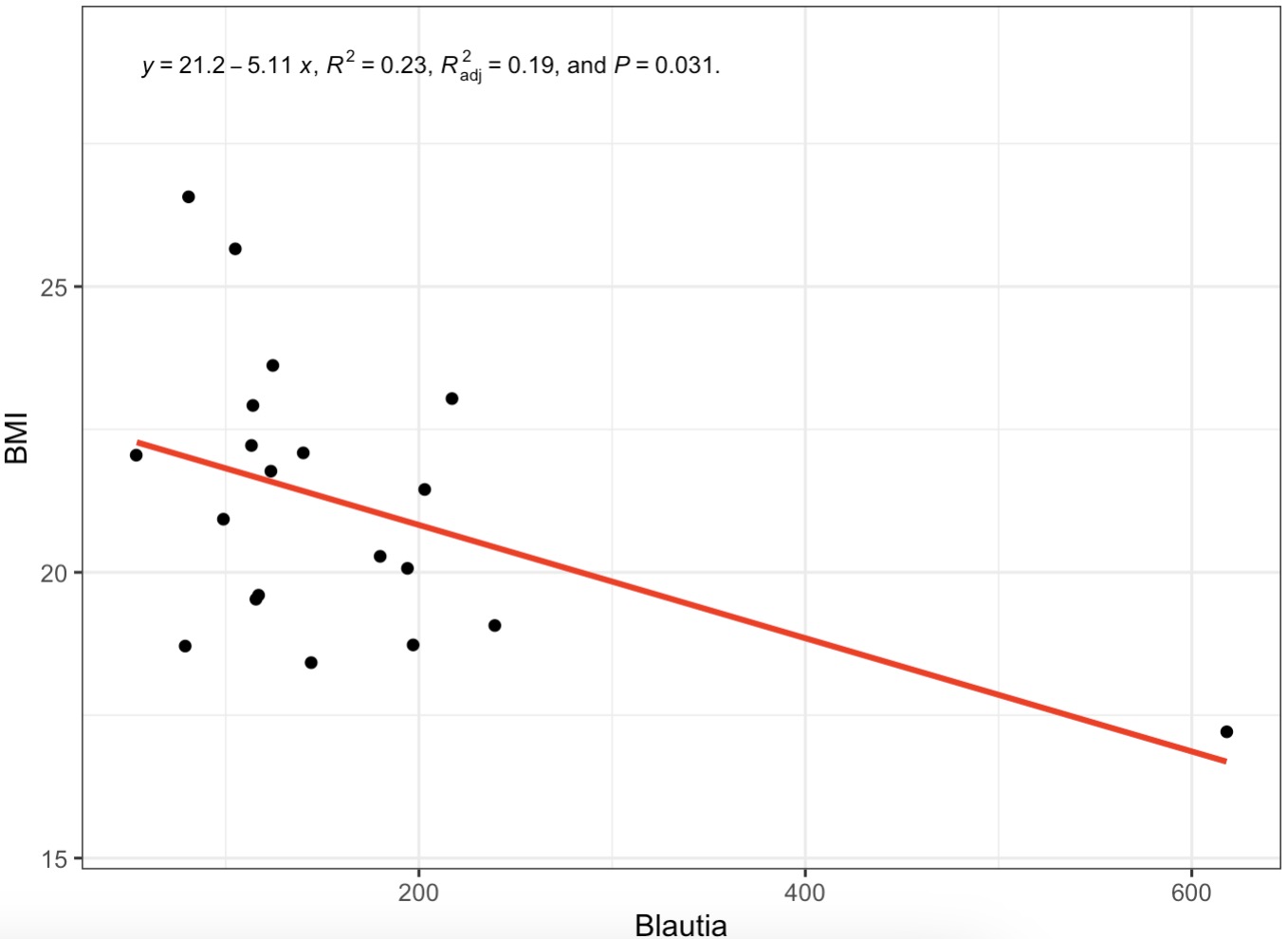
10.6.12 Linear Regression with Phenotypes
Linear Fit Between Genus Blautia and BMI
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'Genus') |>
EMP_fitline_plot(var_select=c('Blautia','BMI'))
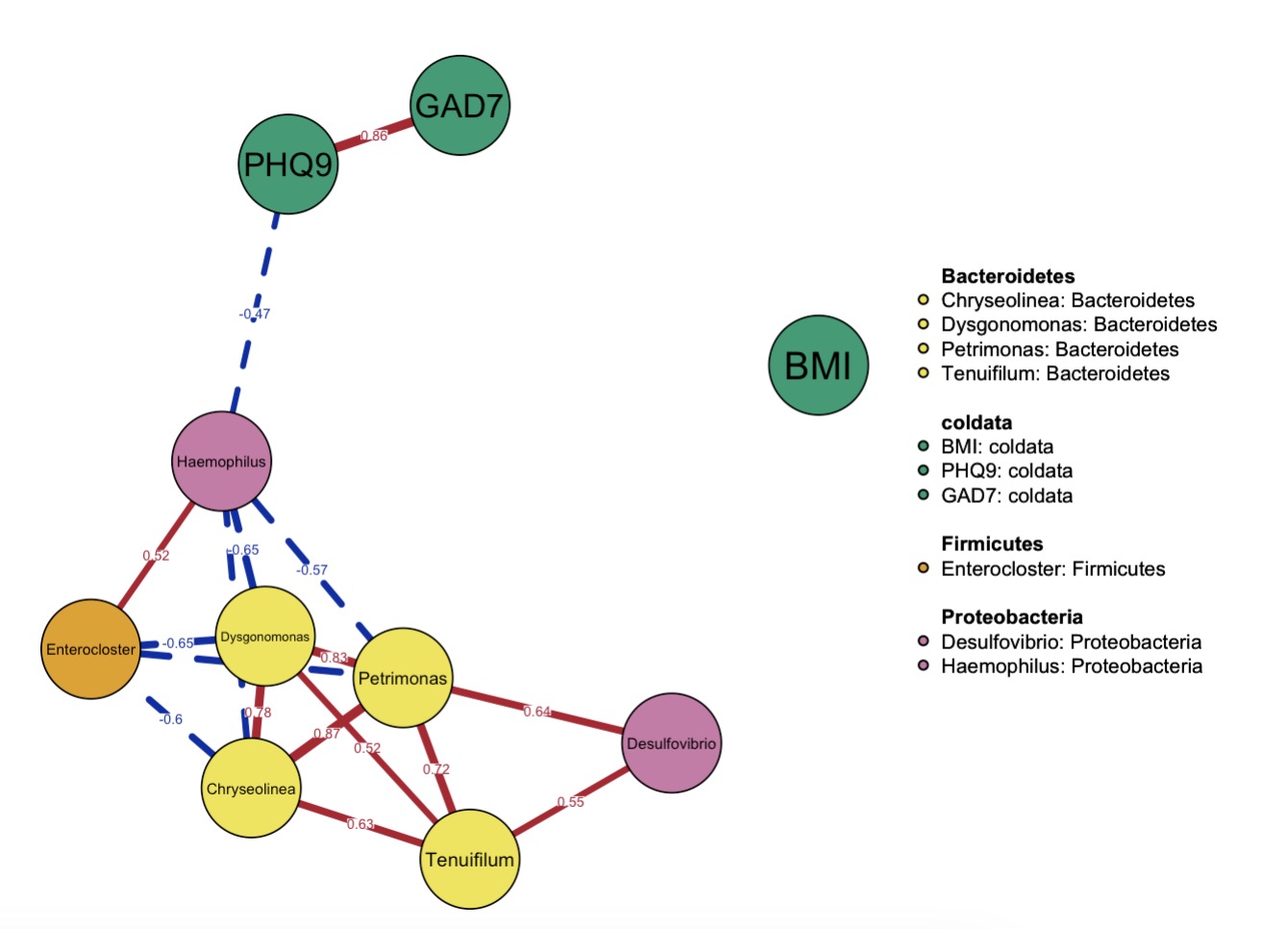
10.6.13 Network Analysis
Network Plot Using Differentially Abundant Genera and Selected Phenotypes
MAE |>
EMP_assay_extract() |>
EMP_collapse(estimate_group = 'Genus',collapse_by = 'row') |>
EMP_diff_analysis(method='wilcox.test', estimate_group = 'Group') |>
EMP_filter(feature_condition = pvalue<0.05) |>
EMP_network_analysis(coldata_to_assay = c('BMI','PHQ9','GAD7')) |>
EMP_network_plot(node_info = 'Phylum',label.cex = 1,edge.labels = TRUE)
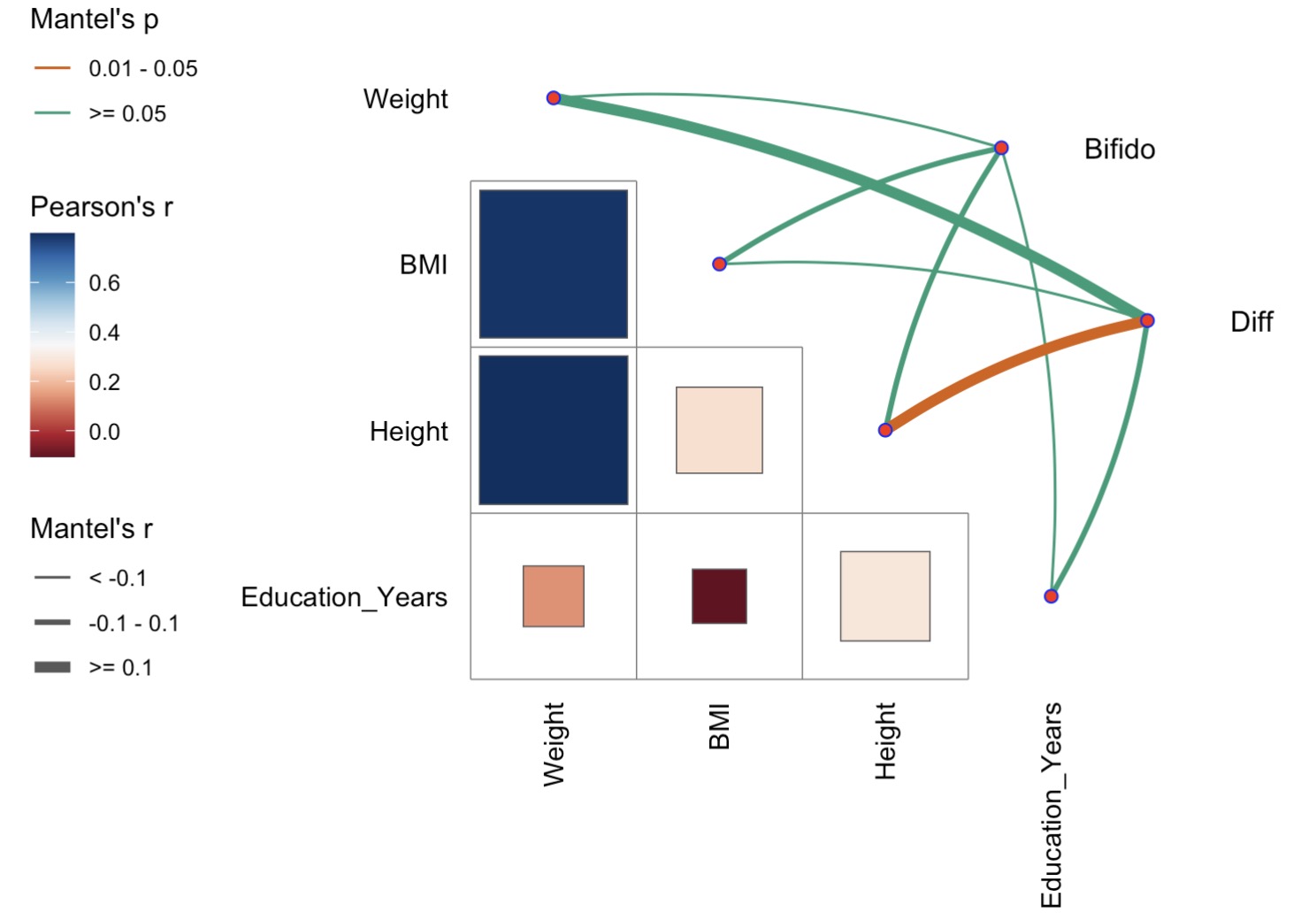
10.6.14 Mantel test
🏷️示例:利用BMI计算差异菌属和双歧杆菌的微生物集与表型数据进行mantel分析
第一步:安装并加载linkET包
devtools::install_github("Hy4m/linkET")
library(linkET)
第二步:利用BMI形成新的分组,并使用oneway差异分析筛选显著差异的菌属
diff_genus <- MAE |>
EMP_assay_extract() |>
EMP_mutate(Degree = dplyr::case_when(
BMI < 18.5 ~ "Lean",
BMI >= 18.5 & BMI < 24 ~ "Normal",
BMI >= 24 & BMI < 28 ~ "Fat",
TRUE ~ "Need Med"
),mutate_by = 'sample',location = 'coldata',.after = Group)|>
EMP_decostand(method = 'relative') |>
EMP_collapse(estimate_group = 'Genus',collapse_by = 'row') |>
EMP_diff_analysis(method = 'oneway.test',estimate_group = 'Degree') |>
EMP_filter(feature_condition = pvalue < 0.05) |>
EMP_assay_extract(action = 'get')
diff_genus
第三步:筛选双歧杆菌的菌属
Bifidobacterium <- MAE2 |>
EMP_assay_extract() |>
EMP_decostand(method = 'relative') |>
EMP_assay_extract(pattern = 'Bifidobacterium',
pattern_ref = 'Genus',action = 'get')
Bifidobacterium
第四步:合并两个微生物数据集并提取出表型数据
tax_data <- purrr::reduce(list(Bifidobacterium,diff_genus),
inner_join,by='primary') |>
tibble::column_to_rownames('primary')
meta_data <- MAE |>
EMP_coldata_extract(action = 'get') |>
dplyr::select(primary,Weight,BMI,Height,Education_Years) |>
tibble::column_to_rownames('primary')
第五步:构建微生物数据和表型数据的mantel分析
mantel <- mantel_test(tax_data, meta_data,
spec_select = list(Bifido = 1:7,
Diff = 8:10)) |>
mutate(rd = cut(r, breaks = c(-Inf, -0.1, 0.1, Inf),
labels = c("< -0.1", "-0.1 - 0.1", ">= 0.1")),
pd = cut(p, breaks = c(-Inf, 0.01, 0.05, Inf),
labels = c("< 0.01", "0.01 - 0.05", ">= 0.05")))
mantel
第六步:绘制mantel可视化结果
qcorrplot(correlate(meta_data,method='pearson'), type = "lower", diag = FALSE) +
geom_square() +
geom_couple(aes(colour = pd, size = rd),
data = mantel,
curvature = nice_curvature()) +
scale_fill_gradientn(colours = RColorBrewer::brewer.pal(11, "RdBu")) +
scale_size_manual(values = c(0.5, 1, 2)) +
scale_colour_manual(values = color_pal(3)) +
guides(size = guide_legend(title = "Mantel's r",
override.aes = list(colour = "grey35"),
order = 2),
colour = guide_legend(title = "Mantel's p",
override.aes = list(size = 2),
order = 1),
fill = guide_colorbar(title = "Pearson's r", order = 2))